Plant Cell Wall Glycobiology


"Plant cell wall degradation"
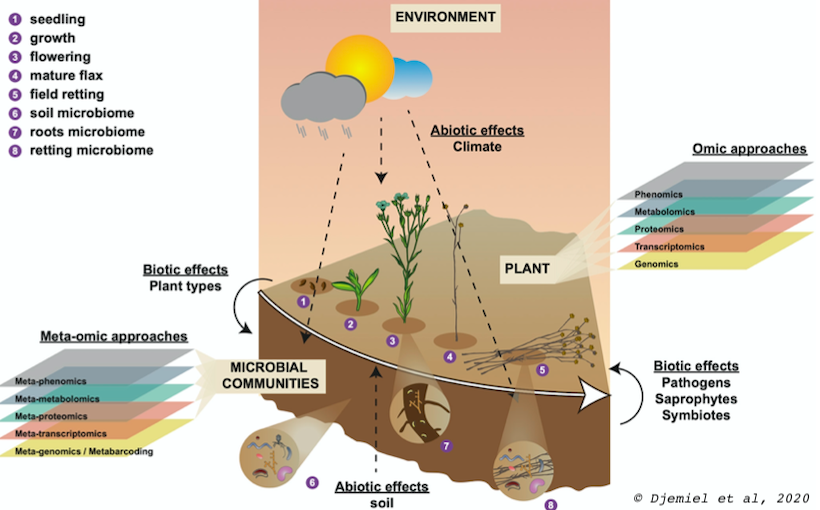
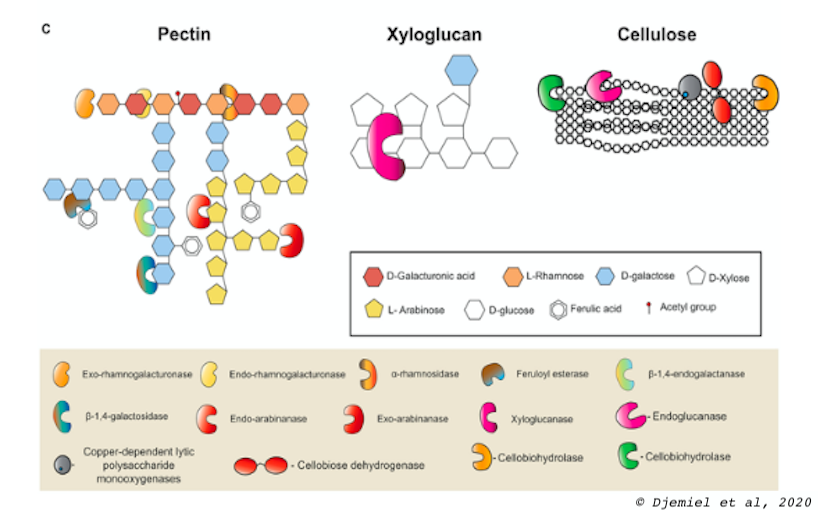
The degradation of the plant wall is a fundamental process allowing the recycling of the biomass produced at the level of the biosphere. It's also an important step into transform of biomass into valuable biopolymers, materials or biofuel. In nature, these processes relie on microbial facilities to secrete cell wall degrading enzymes. Indeed, pathogenic or saprophytic microorganisms possess a large set of secreted enzymes that are dedicated to depolymerization of the main structural polysaccharide components of the plant cell wall, i.e., cellulose, hemicellulose, and pectin. Our laboratory studies the enzymatic functions dedicated to the degradation of polysaccharide polymers of the cell wall.
For that purpose, we coupled metaOmics (metabarcoding, metatranscriptomic and metaproteomics) approach, to classical enzymology and "wet" plant cell wall polysaccharide chemistry to identify some potential major enzymatic functions related to bacterial and fungal cell wall degradation. Classically, this type of approach is carried out on forest beds for which a lot of data already exists. Our originality is that we are interested in a model of controlled natural degradation used in the production of natural fibers: the flax dew retting.
Flax dew retting as a model
Flax dew retting is the first step in the industrial transformation of flax stems into valuable fibers.
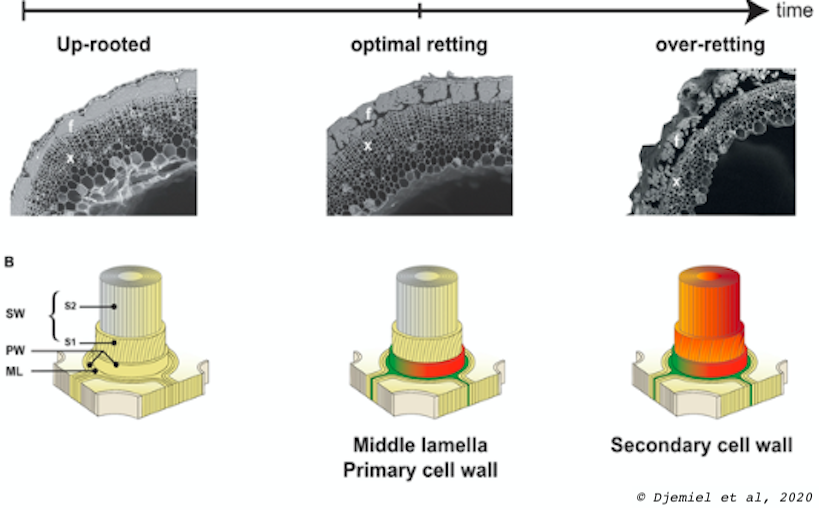
The flax stems are ‘pulled’ (up-rooted) and laid down directly on the soil in swaths (strips). Subsequent exposure to alternating periods of rain and heat combined with the development and the action of soil microflora processes the separation of cellulosic bast fibres from the stems via the partial degradation of cell wall polymers.
Despite many studies of this process relatively little is known about :
i) the composition and the evolution of the microflora population,
ii) the kinetics of microbial colonization of plant material,
iii) the composition and the evolution of the plant cell wall-degrading enzymes targeted to the different CAZy families.


MetaOmics
Thanks to the metaOmics approaches we were able to establish the first comprehensive microbiota of flax dew retting and identify some potential major enzymatic functions related to bacterial and fungal cell wall degradation (Djemiel et al, 2017). These approaches based on Next Generation of Sequencing (Illumina Hiseq and MiSeq system) can access the exogenous (soil) and endogenous (plant) enzymatic arsenal potentially involved in degrading cell wall polysaccharides during dew-retting in flax (Djemiel et al, 2020, Chabbert et al, 2020).
Metaomics represent the high-throughput, global analysis of DNA, RNA, proteins, or metabolites isolated directly from a community of organisms living in a particular environment (Human Genome Project Information Archive Glossary, 2012).
Metagenomics is the study of genetic material recovered directly from environmental samples. The broad field may also be referred to as environmental genomics, ecogenomics or community genomics. While traditional microbiology and microbial genome sequencing and genomics rely upon cultivated clonal cultures, early environmental gene sequencing cloned specific genes (often the 16S rRNA gene) to produce a profile of diversity in a natural sample. Metagenomics allows researchers to access the functional and metabolic diversity of microbial communities, but it cannot show which of these processes are active. The extraction and analysis of metagenomic mRNA (the metatranscriptome) provides information on the regulation and expression profiles of complex communities (Wikipedia 2016).
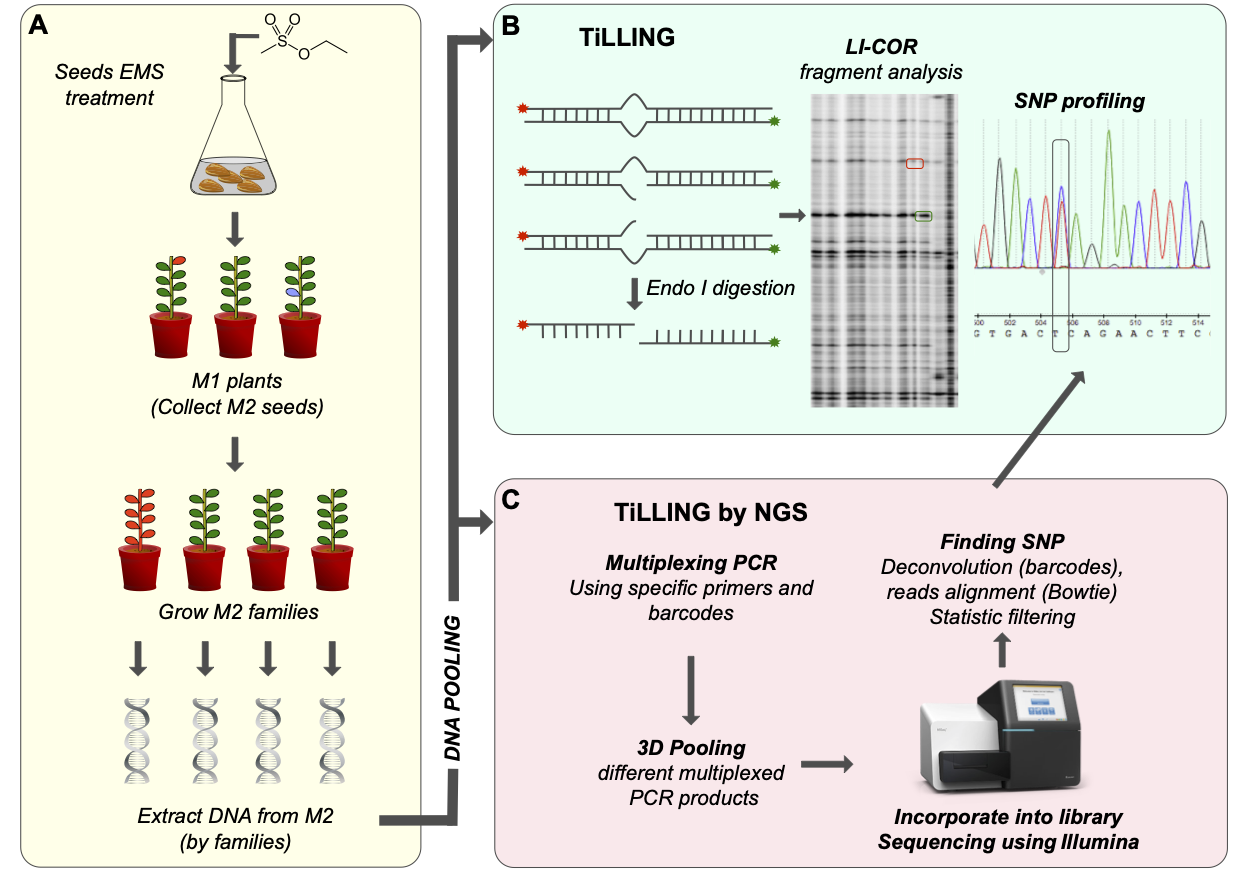
TiLLING approach to study cell wall construction
Project description
The flax genome has been sequenced and transcriptomics are providing information on candidate genes potentially related to agronomically-important traits. In order to accelerate functional characterization of these genes we have generated a flax EMS mutant population that can be used as a TILLinG (Targeting Induced Local Lesions in Genomes) platform for forward and reverse genetics.
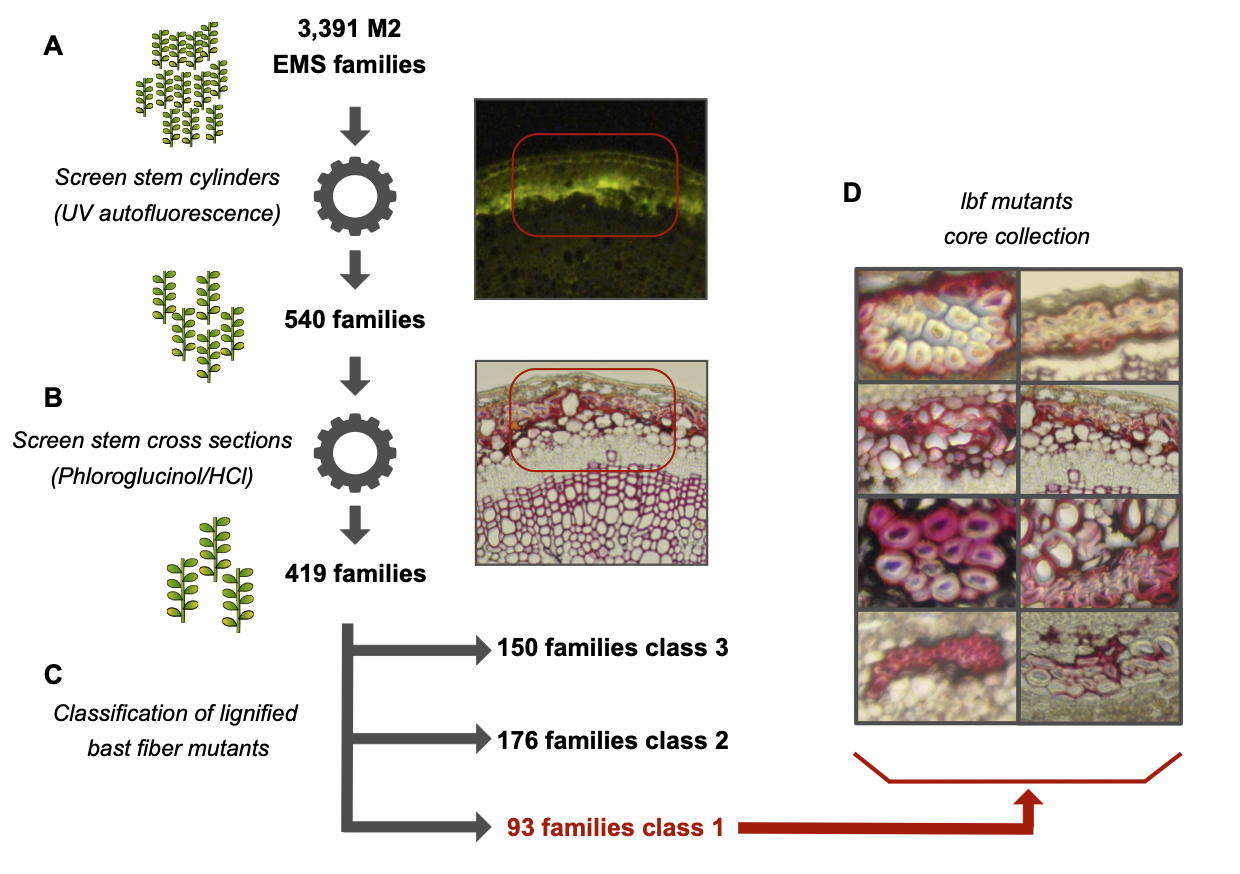
Main results
We have developed a more than 4000 flax mutants population that can be used as an efficient forward and reverse genetics tool. The collection has an extremely high mutation rate that enables the detection of large numbers of independant mutant families by screening a comparatively low number of M2 families. The population will prove to be a valuable resource for both fundamental research and the identification of agronomically-important genes for crop improvement in flax (Chantreau et al, 2013). Moreover, identified mutants producing modified fibers composition leads to a better understand of how fiber are constructed inside flax stem (Chantreau et al, 2014).